
There are 20 unique medications approved for the treatment of HIV infection. Despite this impressive number, there is an indisputable need for new anti-HIV compounds that have potent and durable efficacy profiles, unique resistance patterns, patient-friendly dosing schedules, and minimal toxicities. What follows is an overview of some the newest antiretroviral contenders, discussed by Dr. Roy M. Gulick during a recent meeting of the Physicians’ Research Network.
| I. Nucleoside/Nucleotide Analogues | Top of page |
D-D4FC (Reverset)
Now in phase II clinical trials is D-D4FC (Reverset), a cytidine analogue being developed by Pharmasset Pharmaceuticals and Incyte Corporation. Reverset’s chemical name is D-D4FC and was formerly known as DPC-817 when it was being developed by DuPont Pharmaceuticals and Bristol-Myers Squibb. It has an intracellular triphosphate half-life of 17 hours, rending it suitable for once-daily dosing.
According to encouraging in vitro data, D-D4FC is highly effective in inhibiting subsets of lamivudine-, zidovudine-, and tenofovir-resistant HIV variants but is less potent against variants harboring multiple-NRTI-resistant mutations. D-D4FC selects, in vitro, for the K65R or K70R mutations, which decreases susceptibility to the drug.
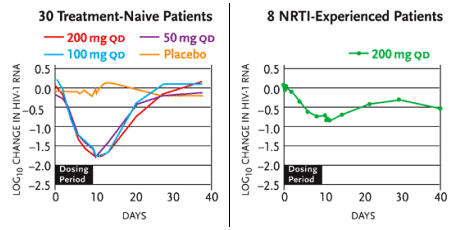
Results of a Phase Ib/IIa study presented by Dr. Robert Murphy of Northwestern University and his colleagues were presented at the XV International AIDS Conference in Bangkok (Murphy, 2004). The placebo-controlled study (Study 202) involved 30 treatment-naïve and 10 treatment-experienced patients. The treatment-naïve patients received 50 mg, 100 mg, or 200 mg D-D4FC monotherapy—or matching placebo—for ten days. Eight treatment-experienced patients, none of whom were maintaining undetectable HIV-RNA levels while taking standard regimens, added 200 mg D-D4FC to their therapies for ten days (and two other others added D-D4FC placebos).
The mean reduction in viral load among the treatment-naïve patients in the three treatment groups ranged from 1.67 log10 copies/mL to 1.77 log10 copies/mL (see Figure 1). Among the treatment-experienced patients, the mean reduction in viral load was 0.8 log10 copies/mL. Seven of eight treatment-naïve patients (in the 200 mg D-D4FC group) and four of eight treatment-experienced patients achieved undetectable viral loads (<400 copies/mL) after ten days of therapy. The drug was well tolerated. No new resistance mutations developed during the ten days of therapy.
Results from a Phase IIb clinical trial (Study 203), involving 180 treatment-experienced patients, are expected soon. Depending on the results of this study, a Phase III clinical trial is planned to begin before the end of 2005.
| Amdoxovir | Top of page |
Amdoxovir (DAPD) is another NRTI that has changed hands several times. It was originally developed by Triangle Pharmaceuticals in collaboration with Emory University and the University of Georgia. In January 2003, Gilead Sciences acquired Triangle Pharmaceuticals and assumed the rights to develop amdoxovir. In March 2004, Gilead Sciences returned the rights to develop amdoxovir to Emory University and the University of Georgia. A new industry sponsor of the compound has not yet been determined.
It is a novel dioxolane purine analogue that is rapidly converted by adenosine deaminase into D-dioxolane guanine (DXG), a metabolite that has potent activity against HIV and HBV. Pharmacokinetics data indicate that it is suitable for twice-daily dosing.
Amdoxovir has antiviral activity, in vitro, against zidovudine- and lamivudine-resistant variants of HIV and is also active against some strains with the 69SSS substitution associated with multiple-NRTI resistance. However, in the presence of amdoxovir and after multiple passages of the virus in vitro, two mutations arise: K65R and L74V.
Of concern has been the toxicity profile of amdoxovir in animal studies and early clinical trials. Long-term toxicology studies indicated that high doses of amdoxovir were associated with lenticular opacities in monkeys and obstructive nephropathy in rats. In one early clinical trial (DAPD-150), no nephrologic toxicities were reported (Thompson, 2003). However, 5/18 (28%) patients in DAPD-150 discontinued the clinical trial because of lens opacities which, fortunately, did not have an impact on visual acuity. It is important to note, though, that on-study assessments were conducted following the animal findings; evaluations at baseline for lens opacities—which can be relatively common—were not performed.
The results of a small clinical trial conducted by the AIDS Clinical Trials Group (A5118) were reported at the 12th Conference on Retroviruses and Opportunistic Infections (CROI) earlier this year in Boston (Gripshover, 2005). This randomized, double-blind, placebo-controlled study evaluated the activity and safety of amdoxovir (300 mg BID) combined with enfuvirtide (Fuzeon) and an optimized background regimen (OBR)—compared to enfuvirtide plus OBR alone—in 18 HIV-infected patients who had failed at least two antiretroviral regimens in the past.
At study entry, the median viral load was 63,000 copies/mL and the median CD4+ count was 36 cells/mm3. The viral resistance profile consisted, on average, of six NRTI and eight protease inhibitor mutations associated with resistance. Thirteen of the 18 patients enrolled also had evidence of non-nucleoside reverse transcriptase inhibitor (NNRTI) resistance).
After 24 weeks of treatment, by the intent-to-treat analysis, there was a 1.1 log10 copies/mL HIV-RNA reduction in the amdoxovir/enfuvirtide/OBR group, compared to a 0.8 log 10 copies/mL reduction in the placebo/enfuvirtide/OBR group. The difference between the two groups was not statistically significant. Similarly, there was no statistically significant difference between the two groups with respect to CD4+ cell count increases (70 cells/mm3 vs. 54 cells/mm3 respectively) or adverse events.
“The ACTG investigators looked carefully for lens toxicities and didn’t find any,” Dr. Gulick commented. “The conclusion of this study was that that compound appeared generally safe and tolerable. Unfortunately, it wasn’t powered sufficiently to show an antiviral effect. There are additional plans to develop this compound, with an emphasis on patients with nucleoside analogue resistance.”
Nucleseoside-Competing Reverse Transcriptase Inhibitors (NcRTIs)
Investigators associated with Tibotec Pharmaceuticals have reported on the in vitro activity of a new compound, dubbed compound X, representing an investigational class of reverse transcriptase inhibitors that are believed to be different from current NRTIs. Although structurally unrelated to nucleosides (or nucleotides) these novel NRTIs nonetheless competitively inhibit reverse transcriptase, hence the designation NcRTIs: nucleotide-competing reverse transcriptase inhibitors.
“Compound X has properties of a nucleoside analogue with a non-nucleoside structure,” Dr. Gulick said. “It’s highly potent in vitro and test tube studies show that this new compound is active against nucleoside and non-nucleoside-resistant viruses.” According to some preliminary data reported at the 12th CROI, compound X is active against variants harboring the Q151M mutation, as well as the 69SSS insertion (Jochmans, 2005). “There’s no clinical candidate in this class yet, but it certainly is interesting to see that there’s a new mechanism by which the reverse transcriptase enzyme can be inhibited.”
| II. New Non-Nucleoside Reverse Transcriptase Inhibitors | Top of page |
Capravirine
The first NNRTI reviewed by Dr. Gulick was Pfizer’s capravirine. In vitro studies have demonstrated that capravirine is active against HIV isolates containing single reverse transcriptase substitutions such as K103N, V106A, and L100I—three mutations that confer resistance to other NNRTIs. However, HIV with dual mutations at positions 100 and 103 resulted in a 24- to 40-fold decrease in sensitivity. The common single Y181C mutation also decreased susceptibility to capravirine by 13-fold (Potts, 1999).
The clinical development of capravirine has been a series of fits of starts. The drug was originally purchased by Agouron PharmaceuticalsThe first NNRTI reviewed by Dr. Gulick was Pfizer’s capravirine. In vitro studies have demonstrated that capravirine is active against HIV isolates containing single reverse transcriptase substitutions such as K103N, V106A, and L100I—three mutations that confer resistance to other NNRTIs. However, HIV with dual mutations at positions 100 and 103 resulted in a 24- to 40-fold decrease in sensitivity. The common single Y181C mutation also decreased susceptibility to capravirine by 13-fold (Potts, 1999).
The clinical development of capravirine has been a series of fits and starts. Agouron Pharmaceuticals originally purchased the drug from Japan Tobacco, Inc. in 1995. Four years later, the development of capravirine seemed uncertain, after the purchase of Agouron Pharmaceuticals by Warner-Lambert. Pfizer merged with Warner-Lambert in 2000, further shaking up plans for capravirine. There have also been regulatory concerns. In early 2001, the FDA and Pfizer announced that capravirine use in clinical trials would be restricted because of animal toxicology studies demonstrating unexpected vasculitis in dogs. However, the capravirine dose associated with vasculitis was significantly higher than the dose currently being studied in humans, and no cases of vasculitis have been detected in patients participating in clinical trials. In December 2001, the FDA took capravirine off clinical hold, and studies have since resumed.
As for the potential efficacy of capravirine, one phase I trial reported that the drug is roughly ten times more potent than any of the current NNRTIs (Hernandez, 2000). Used as monotherapy in treatment-naïve patients, capravirine (2,100 mg BID) resulted in an HIV-RNA reduction of 1.7 log10 copies/mL after ten days of treatment.
Preliminary results from a phase II clinical trial of capravirine involving 75 NNRTI-experienced patients were presented four years ago at the 8th CROI in Chicago (Wolfe, 2001). The study compared two doses of capravirine—1,400 mg BID and 2,100 mg BID—to a placebo, with all three groups of patients receiving nelfinavir and two new NRTIs. Approximately 25/50 (50%) evaluable patients who received either dose of capravirine had HIV-RNA levels below 400 copies/mL after 12 weeks of treatment. Among the 12 patients who had been receiving treatment for 16 weeks in the placebo group, HIV-RNA levels had decreased by 1.5 log copies/mL. Among the eight evaluable patients in the 1,400 mg capravirine group, the median HIV-RNA decrease after 16 weeks was 2.2 log copies/mL. As for the 10 evaluable patients in the 2,100 mg capravirine group, the median viral load decrease was 1.7 log copies/mL after 16 weeks of treatment. In terms of adverse events, diarrhea, nausea, and vomiting occurred more frequently in the 2,100 mg group than in the 1,400 mg or placebo groups. At the time of this presentation, four patients had discontinued because of treatment failure and seven patients had discontinued because of adverse events.
The most recent Phase II clinical trial results to be presented come from Study 1002 (Pesano, 2005). The study enrolled 198 protease inhibitor-naïve patients who had failed an NNRTI-based regimen and randomized them to receive capravirine, either 700 mg or 1,400 mg BID, or placebo, plus nelfinavir (Viracept) and two NRTIs (based on treatment history and genotyping results). At baseline, the median viral load was 4.4 log10 copies/mL and the average CD4+ count in the three study groups was between 200 and 250 cells/mm3. Resistance to NRTIs and NNRTIs was similar across the study arms.
Reported failure rates at 48 weeks—defined as failure to reduce HIV-RNA levels by 0.5 log10 copies/mL by Week 4 (or a viral load rebound after an initial reduction of at least 0.5 log10 copies/mL)—were 24% in the placebo group, 15% in the 700 mg capravirine group, and 13% in the 1,400 mg capravirine group. These differences were not statistically significant. Also falling short of statistical significance were the percentages of patients in each group with HIV-RNA levels below 50 copies/mL: 40% in the capravirine 700 mg group, 47% in the capravirine 1,400 mg group, and 39% in the placebo group. No differences in CD4+ cell count gains or adverse events were noted.
“The conclusion of this study,” Dr. Gulick noted, “was that capravirine did not seem to add appreciable antiretroviral activity in this patient population. Part of the difficulty we have with interpreting these data is that patients were protease inhibitor-naïve at baseline and everyone in the study received a protease inhibitor and new nucleoside analogues. It is possible the HIV-RNA effect was dampened.” Nonetheless, additional capravirine studies are planned, including a clinical trial that will enroll patients with triple-class failure to receive lopinavir/ritonavir (Kaletra) plus capravirine.
TMC125
Tibotec’s TMC125 represents a new class of NNRTI known as diaminopyrimidine (DAPY) compounds. They have a flexible structure that, at least in vitro, has activity against both wild-type HIV strains and those containing key mutations associated with NNRTI resistance. “The three approved NNRTIs bind tightly in the active site of HIV’s reverse transcriptase and can’t tolerate amino acid side chain substitutions in the binding site,” Dr. Gulick explained. “These newer NNRTIs are more flexible. That is, they may be able to tolerate mutations within the active site of the enzyme.”
In antiretroviral-naive patients, seven days of TMC125 monotherapy resulted in a 1.99 log10 copies/mL reduction in HIV-RNA (Gruzdev, 2003). In fact, data presented at the 9th CROI suggested that the drug—as monotherapy—results in a similar initial rate of decline of HIV-RNA during the first week of treatment as a five-drug, PI- and NNRTI-containing regimen (Sankasing, 2002).
In vitro data evaluating the effectiveness of TMC125 against a panel of NNRTI-resistant viruses were reported at the 11th CROI in San Francisco (Vingerhoets, 2004). To put these data into context, the investigators established that the IC50 for TMC125 against wild-type virus was 0.9 nM. Three of 54 variants harboring single mutations known to confer at least partial resistance to the current crop of NNRTIs—Y181I, Y181V, and F227C—had a greater than tenfold reduction in sensitivity to TMC125. Of 19 variants harboring two mutations, one strain—containing V179F and Y181C—resulted in a greater than tenfold reduction in TMC125 sensitivity. More prevalent combinations of two NNRTI mutations—including L100I, K103N, Y181C, and G190A—all resulted in less than a tenfold decrease in TMC125 sensitivity. Conversely, of variants containing two of these key mutations, 11 of 19 had greater than tenfold reduction, and 4 of 19 had greater than hundredfold reductions, in sensitivity to efavirenz. As for variants harboring three key NNRTI-associated mutations—a rare occurrence in the real world, say researchers at Virco—decreased sensitivity to TMC125 was observed, albeit to a lesser degree than efavirenz.
Data from a study evaluating the short-term effects of TMC125 in NNRTI-experienced patients with high levels of drug resistance to currently available NNRTIs have been published (Gazzard, 2003). Sixteen patients, all of whom had between 10- and 500-fold resistance to either efavirenz (Sustiva) or nevirapine (Viramune), switched their faltering NNRTI for nine 100 mg twice-daily TMC125 capsules (900 mg BID) for seven days. Twelve of the patients enrolled in this study had at least two reverse transcriptase mutations that conferred high-level resistance to both efavirenz and nevirapine. On day 8, the average decrease in HIV-RNA was 0.86 log10 copies/mL, with 12 patients achieving a greater than 0.5 log reduction and seven patients achieving a greater than 1 log decline. Interestingly, no association was apparent between the observed antiviral responses and baseline NNRTI resistance. Tolerability was also reported to be good, with mild headaches and diarrhea—possibly contributed to by the inactive ingredients in the current capsule formulation—being the most common side effects reported. Overall, these results demonstrated the short-term activity of TMC-125 in NNRTI-experienced patients, albeit to a comparatively lesser degree than those seen in NNRTI-naïve patients.
TMC 278
Another DAPY compound slated for development is Tibotec’s TMC 278. “This is a small molecule, which may have one key advantage,” Dr. Gulick said. “The smaller the molecule, the more potential it has in terms of being coformulated in a single pill with other antiretrovirals. That’s the impetus behind developing this compound.”
According to preliminary data presented at the 12th CROI, TMC 278 was active against 89% of 1,500 NNRTI-resistant HIV isolates, compared to 33% using efavirenz (Sustiva) and 0% using nevirapine (de Bethune, 2005). Using higher concentrations of the drug, no NNRTI mutations were documented in vitro. However, with lower concentrations of the drug, as many as eight mutations emerged, including L100I, V106I, Y181C, and M230I. Pharmacokinetic data indicate that the compound is well absorbed and has a half-life ranging from 34 to 55 hours: suitable for once-daily—or even less frequent—dosing.
Preliminary safety and efficacy data from a Phase IIa placebo-controlled study were also reported at the 12th CROI (Goebel, 2005). The 47 patients enrolled were treatment naïve, had viral loads in excess of 5,000 copies/mL, a CD4+ count between 75 cells/mm3 and 500 cells/mm3, and no evidence of antiretroviral drug resistance. Patients received either 25 mg, 50 mg, 100 mg, or 150 mg TMC 278, via oral solution once daily, or placebo for seven days.
After seven days of treatment, the median HIV-RNA drop was 1.2 log10 copies/mL; the antiviral effect was similar in all four dosing groups. The most common adverse effect in the placebo and treatment groups was headache. Grade 1 rash occurred in one patient receiving TMC 278; Grade 3 nausea occurred in another patient receiving TMC 278. No NNRTI mutations emerged between baseline and the end of treatment.
Other studies of TMC 278 are being carried out in treatment-experienced patients.
| III. Protease Inhibitors | Top of page |
Tipranavir (Aptivus)
Tipranavir (Aptivus) is a nonpeptidic dihydropyrone, a new class of protease inhibitors believed to have greater flexibility in conforming to enzyme variants resistant to current protease inhibitors. The compound was originally developed by Pharmacia & Upjohn and is now gearing up for approval as a Boehringer Ingelheim product.
In addition to its unique chemical structure, tipranavir also differs from other currently available protease inhibitors in its metabolic profile. The drug induces the cytochrome P450 pathway, whereas current protease inhibitors either inhibit or both inhibit and induce this enzyme system. In early phase II studies, 1,500 mg of tipranavir, taken three times daily, was required to achieve the necessary trough concentration. To circumvent this hurdle, the manufacturer developed a self-emulsifying drug delivery system (SEDDS)—a soft-gel capsule—for the compound. After taking the new formulation of tipranavir into a phase II study (Study 1182.52), the manufacturer concluded that the optimal tipranavir dose is 500 mg twice daily and will need to be combined with low doses of ritonavir (200 mg twice daily) to reverse the rapid metabolism of the drug by cytochrome P450 and to allow dosing with food (Gathe, 2003).
Study 1182.52 also produced some important resistance data (Cooper, 2003). In this study, patients who had tried at least two protease inhibitors in the past and had strains of HIV harboring at least one common protease-associated mutation (PRAM)—33I/V/F, 82A/F/L/T, 84V, and/or 90M—were randomized to receive one of three tipranavir doses in combination with ritonavir (see dose-optimization conclusions above). According to phenotypic analyses of 157 isolates collected at the start of the study (216 patients were enrolled), the median fold increases in IC50 ranged from 7.0 to 94.2 for all of the currently approved protease inhibitors, compared to a 1.1-fold increase in the tipranavir IC50 against these highly resistant isolates. Tipranavir’s IC50 increase was onefold or less in 42% of the isolates, between onefold and twofold in 27% of the isolates, between twofold and fourfold in 18%, and greater than fourfold in 12%. Among patients harboring HIV strains with twofold or less resistance to tipranavir, viral load decreased, on average, by 1.23 log10 copies/mL during the first month of the study. Among patients with greater than twofold resistance to tipranavir, median viral load decreases were less than 0.25 log10 copies/mL. In other words, a greater than twofold increase in tipranavir’s IC50 was a breakpoint for the drug. However, the investigators noted that an accumulation of a large number of protease gene mutations was necessary to cause a significantly diminished antiviral response to tipranavir. Patients with three or more PRAMs were not likely to benefit from tipranavir therapy. However, data from subsequent studies determined that this was a flawed conclusion.
One of the subsequent studies of interest is Study 1182.51 (Leith, 2004). This open-label study evaluated the pharmacokinetics of tipranavir/ritonavir alone and in combination with saquinavir, amprenavir, or lopinavir/ritonavir (Kaletra) in 315 highly treatment-experienced patients with three or more PRAMs prior to study entry. Patients optimized their background regimens on the basis of resistance testing and added: 1) tipranavir/ritonavir 500 mg/200 mg twice daily; 2) saquinavir/ritonavir 1000 mg/100 mg twice daily; 3) amprenavir/ritonavir 600 mg/100 mg twice daily; or 4) lopinavir/ritonavir 400 mg/100 mg twice daily. After two weeks, tipranavir/ritonavir 500 mg/100 mg twice daily was added to the three groups that didn’t initially receive tipranavir. Minimum concentrations (Cmin) were evaluated at the end of weeks 1, 2, 3, and 4, and a 12-hour intensive pharmacokinetics evaluation was conducted both before and after addition of tipranavir to the other protease inhibitors.
Because of its unique metabolic effects, tipranavir reduced the Cmin values of amprenavir, saquinavir, and lopinavir by 56%, 55%, and 81% respectively. As a result, after four weeks of double-boosted PI therapy, HIV-RNA values rose back to baseline values in all study groups. Based on these results, and until double-boosted PI optimization studies are conducted, it is not recommended that tipranavir be used in combination with any other PIs, with the exception of ritonavir.
Nonetheless, the initial viral load drop of approximately 1.2 log10 copies/mL in these heavily pre-treated patients was encouraging. “The patients’ HIV-RNA levels started to rebound after several weeks on therapy,” Dr. Gulick added. “However, this shouldn’t come as a surprise, given that many patients didn’t have other effective agents to pair with tipranavir. The bottom line from this study was that there was demonstrated activity in these patients using tipranavir, even though they were highly resistant to available protease inhibitors.”
Twenty-four-week results from the Phase III RESIST studies have been reported over the past year. RESIST-1 (BI1182.12) is being conducted in the United States, Canada, and Australia. RESIST-2 (BI1182.48) is being conducted in Europe and Latin America. At study entry, patients were required to have used at least two protease inhibitor-inclusive regimens in the past and have a viral load of at least 1,000 copies/mL. Patients were also required to have an HIV variant harboring at least one primary mutation conferring decreased sensitivity to protease inhibitors (30N, 46I/L, 48V, 50V, 82A/F/L/T, 84V, or 90M). However, the study participants were not allowed to have any more than two PRAMS (33I/V/F, 8A/2F/L/T, 84V, and/or 90M). In both studies, patients were randomized to receive tipranavir/ritonavir or a ritonavir-boosted comparator protease inhibitor (CPI/r) regimen (amprenavir, indinavir, lopinavir, or saquinavir combined with ritonavir).
In RESIST-1, the treatment response after 24 weeks, defined as a 1 log10 or greater decrease in viral load from baseline, was achieved by 41.5% of patients who received tipranavir/r, compared to 22.3% of patients in the CPI/r arm (Hicks, 2004). Viral load reductions were more pronounced in the tipranavir/r group compared to the CPI/r group after 24 weeks (-0.88 vs. -0.28 log10 copies/mL respectively) and more patients in the tipranavir/r group were likely to have HIV-RNA levels below 50 copies/mL compared to those in the CPI/r group (25.1% vs. 10% respectively). All of these reported differences were statistically significant.
Patients receiving tipranavir/r in RESIST-1 also experienced greater increases in their CD4+ cell count than those receiving CPI/r, with CD4+ count increases of 36 cells/mm3 and 6 cells/mm3 respectively.
Results of RESIST-2 were similar to those of RESIST-1. The treatment response after 24 weeks, again defined as a 1 log10 or greater decrease in viral load from baseline, was achieved by 41% of patients who received tipranavir/r, compared to 14.9% of patients treated in the CPI/r group (Cahn, 2004). A greater proportion of patients receiving tipranavir/r achieved a viral load below detectable limits than those who were treated with a CPI/r. At 24 weeks, 22.5% of participants in the tipranavir/r group and 8.6% of participants in the CPI/r group achieved viral loads of less than 50 copies/mL. Patients taking tipranavir/r also experienced greater increases in their CD4+ counts than those in the CPI/r group, with CD4+ count increases of 31 cells/mm3 and 1 cell/mm3 respectively.
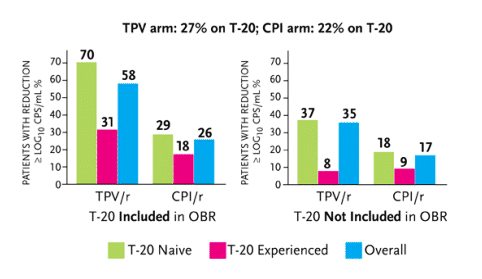
An analysis of enfuvirtide coadministration with tipranavir/r in the RESIST studies was reported at the 12th CROI (Cooper, 2005). In both RESIST studies, patients receiving enfuvirtide (Fuzeon) as part of their treatment are generally more immune-compromised. In RESIST-1, 36.1% of patients used enfuvirtide; in RESIST-2, only 11% used enfuvirtide. As described in Figure 2, 27.1% of patients receiving tipranavir/r in both studies used enfuvirtide, compared to 22.2% of patients in both studies receiving CPI/r. Among patients receiving tipranavir/r, 30% also receiving enfuvirtide had HIV-RNA levels below 50 copies/mL after 24 weeks, compared to 24% receiving tipranavir/r without enfuvirtide. And among patients receiving CPI/r, 13% had HIV-RNA levels below 50 copies/mL after 24 weeks, compared to 10% receiving CPI/r without enfuvirtide, although these data were not statistically significant.
A detailed resistance analysis of pooled RESIST data was also presented at the 12th CROI (Schapiro, 2005). The patients in the RESIST studies had received an average of 12 antiretroviral agents prior to enrollment, with 12% receiving enfuvirtide either prior to study entry or at the time of enrollment (some patients continued enfuvirtide therapy into the RESIST studies).
Among patients with any mutations associated with protease inhibitor resistance, the virologic response to tipranavir/r was better than the response in the CPI/r group. Approximately 50% of patients with 12 or fewer protease mutations at baseline receiving tipranavir/r had a 1 log10 copies/mL or greater decrease in HIV-RNA, compared to 29.8% of patients in the CPI/r group. Approximately 39% of patients receiving tipranavir/r with 13 to 15 protease mutations had a 1 log10 or greater HIV-RNA reduction, compared to approximately 26% of patients in the CPI/r group. Among patients with 16 to 18 protease mutations at baseline, 43.6% of the tipranavir/r group versus 13% of the CPI/r group had a l log10 copies/mL or greater decrease in their viral loads after 24 weeks. And among those with 19 or more mutations, 31.7% in the tipranavir/r group vs. 7.7% in the CPI/r group had a 1 log10 copies/mL or greater decrease in HIV-RNA.
Also analyzed was the number of primary protease inhibitor mutations in relation to tipranavir/r and CPI/r susceptibility. Among patients with one or two primary protease inhibitor mutations, 40.9% of patients in the tipranavir/r group and 28% of patients in the CPI/r group had a 1 log10 copies/mL or greater reduction in HIV-RNA. Approximately 42% of patients receiving tipranavir/r, compared to 13.6% of patients receiving CPI/r, with three or four primary protease inhibitor mutations had a 1 log10 copies/mL or greater reduction in HIV-RNA. Similarly, among patients with five to six primary protease inhibitor mutations, 44.4% in the tipranavir/r group and 16.7% in the CPI/r group had HIV-RNA reductions of 1 log10 copies/mL or greater after 24 weeks of treatment.
Finally, in evaluating the relationship between the number of PRAMs and virologic responses in the RESIST studies, it was noted that patients receiving tipranavir/r had more robust virologic responses than patients receiving CPI/r, regardless of the number of PRAMs. “The bottom line here was that a tipranavir mutational score that comprised mutations at 16 different locations in the protease gene, for a total of 21 total mutations, was the most accurate way to predict tipranavir resistance,” Dr. Gulick summarized. “PRAMs do not accurately predict virologic response to this drug.”
TMC114
Much like its NNRTI contender TMC 125 (discussed above), Tibotec dubs TMC114—its lead protease inhibitor candidate—a “resistant-repellent” compound. More specifically, TMC114 was designed not only to bind with high affinity to typical active sites of the protease enzyme, but also to remain active because of its unique flexibility in the event of mutations that arise during prior therapy with other protease inhibitors.
A dose-escalating study of TMC114 has been conducted (Van der Geest, 2001). Two groups of nine HIV-negative volunteers (six active, three placebo) received alternating doses of 100 mg, 200 mg, 400 mg, 800 mg, 1,200 mg, or 1,600 mg. Because the maximum tolerated dose was not reached, an additional panel was added to receive 2,400, 3,200 and 4,000 mg. Initially, plasma concentrations increased more than proportionally with the dose. No further increases in plasma concentrations were observed between 2,400 mg and 3,200 mg. The mean Cmax was 14.4–15.3 mg/ml at these dose levels. The elimination half-life was approximately 10 hours, irrespective of dose. For 800 mg doses and higher, plasma levels at eight to 12 hours post-dose exceeded protein-adjusted IC50s for isolates resistant to currently approved protease inhibitors. All doses were considered safe. Diarrhea—because of polyethylene glycol (PEG) in the formulation—occurred at high dose levels and limited further escalation. Short-term localized oral and peripheral paresthesias were observed in three of six volunteers receiving the 3,200 mg TMC114 dose.
In a phase II clinical trial reported at the 11th CROI, 50 patients failing a protease inhibitor-based regimen—and a history of other protease inhibitor failures in the past—were randomized to switch their current protease inhibitor to one of three doses of TMC114 (combined with 100 mg ritonavir) or to continue their failing regimen (Peeters, 2004). At baseline, patients had a median viral load of 4.3 log10 copies/mL and 46% were resistant to all of the currently approved protease inhibitors. After 14 days, the patients who did not switch their protease inhibitor(s) for TMC114 experienced a slight increase in their viral loads. Those in the 300 mg, 600 mg, and 900 mg TMC114 groups had median HIV-RNA decreases of 1.24 log10, 1.5 log10 and 1.3 log10 copies/mL respectively. No patients in the control group achieved undetectable viral load levels (<400 copies/mL); after 14 days of treatment, in 31%, 42% and 46% of those in the 300 mg, 600 mg, and 900 mg TMC114 groups achieved viral loads <400 copies/mL. Importantly, there was no correlation between baseline resistance and virologic outcome.
Additional Phase II clinical trial results, from a planned interim 24-week analysis of Study C213 and Study C202, were reported at the 12th CROI (Katlama, 2005). These studies enrolled 397 triple-class experienced patients—all of whom had one or more primary protease inhibitor mutations, a median baseline viral load of 4.51 log10 copies/mL, and a median CD4+ count of 136 cells/mm3—to receive TMC/r (400/100mg or 800/100 mg QD, or 400/100 mg or 600/100 mg BID) or a comparative protease inhibitor plus ritonavir (CPI/r). All patients also received an optimized background regimen, based on the results of genotypic testing.
Figure 3. TMC-114 Study: Subset Analysis (Viral Load <50 Copies/mL at Week 24)
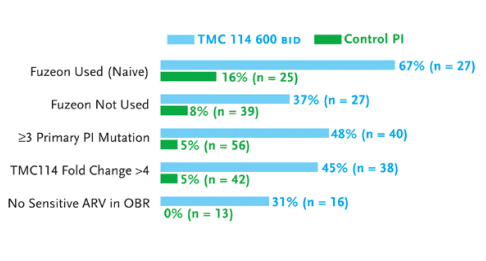
Source: Katlama, 2005
Results were most favorable among patients receiving the highest dose of TMC-114/r (600/100 mg BID). The median change in viral load, after 24 weeks, was -1.85 log10 in the highest dose TMC-114/r group, compared to -1.3 to -1.5 log10 in the other TMC-114/r groups and -0.27 log10 in the CPI/r groups. Also after 24 weeks, 47% in the highest dose TMC-114/r group had HIV-RNA levels below 50 copies/mL, compared to 30% to 38% in the other TMC-114 groups and 9% in the CPI/r groups. CD4+ counts increased by 75 cells/mm3 in the highest dose TMC-114/r group, compared to an increase of 15 cells/mm3 in the CPI/r groups.
With enfuvirtide as a new agent combined with TMC-114/r, 67% in the highest dose TMC-114/r group had HIV-RNA levels below 50 copies/mL (n=27), compared to 16% of patients in the CPI/r groups (n=25)(see Figure 3). Among patients with three or more primary protease inhibitor mutations, 48% in the highest dose TMC-114/r group (n=40), compared to 5% in the CPI/r groups (n=56), had HIV-RNA levels below 50 copies/mL after 24 weeks. And among patients who had no antiretrovirals in the optimized background regimen to which their HIV was sensitive, 31% in the highest dose TMC-114/r group (n=16), compared to 0% of the patients in the CPI/r group (n=13), had HIV-RNA levels below 50 copies/mL after 24 weeks.
“These are impressive results that we really haven’t seen before in a highly treatment-experienced patient group,” Dr. Gulick optimistically noted.
| IV. Entry and Fusion Inhibitors | Top of page |
Maraviroc
UK-427,857, which now carries the generic name maraviroc, is a chemokine receptor antagonist designed to block the CCR5 receptor. The drug is being developed by Pfizer Pharmaceuticals.
Some preliminary pharmacokinetics data are available (Muirhead, 2005). Maraviroc is a CYP 3A4 substrate, not an inhibitor. In single dose studies in HIV-infected individuals, efavirenz decreased maraviroc by 40% to 50%, whereas nevirapine increased the maraviroc Cmax 1.5 times, with little effect on the maraviroc AUC. Lopinavir/r also increased the maraviroc Cmax approximately 1.8 times.
Preliminary results of a phase I study were presented at the 43rd Interscience Conference on Antimicrobial Agents and Chemotherapy in 2003 (Pozniak, 2003). In this study, 24 HIV-positive individuals with CCR5-tropic virus were randomized to receive once-daily oral doses maraviroc (25 mg QD or 100 mg BID) or placebo monotherapy for 14 days. Steady-state drug levels were achieved after seven days, with the best plasma concentrations achieved when taken in the fasted state. After 14 days, an average 1.4 log10 copies/mL reduction in HIV-RNA was seen in patients receiving maraviroc 100 mg BID, compared to a 0.4 log10 copies/mL reduction among patients receiving the 25 mg QD dose. The drug was well tolerated; no serious adverse events were reported.
The preliminary results of a dose-ranging study were reported at the XV International AIDS Conference in Bangkok (Fätkenheuer, 2004). In this study, 80 patients infected with CCR5-tropic HIV received one of several doses of maraviroc (25, 100, or 300 mg QD; or 50, 100, 150, or 300 mg BID) or placebo for a 10-day period. The agent was well tolerated and caused dose-related reductions in viral load. Using the 100 and 300 mg BID dosing, viral load was reduced by 1.4 to 1.6 log10 copies/mL. Food intake did not affect drug levels or antiviral activity.
Also of interest, the study noted that the CCR5 receptor was saturated by at least 85% with all but the lowest dose studied. With respect to tropism, 61/63 evaluable patients remained CCR5 tropic throughout the 10-day treatment course and throughout the additional 30-day follow-up period. Two patients became dual tropic. One reverted back to CCR5 at day 40, whereas the other patient continued to have a dual-tropic HIV genotype without obvious clinical progression after six months.
Vicriviroc
Vicriviroc is the tentative generic name for Schering-Plough’s CCR5 receptor antagonist, also known as SCH 417690 and formerly known as SCH-D. Like maraviroc, the drug is orally bioavailable. It has a long half-life allowing for once-daily dosing. Concentrations of vicriviroc are enhanced with ritonavir, whereas concentrations are decreased with efavirenz or nevirapine.
Phase I clinical trial results, involving 48 HIV-infected patients with CCR5-tropic virus receiving various doses of the drug, were reported at the 11th CROI (Schurmann, 2004). Sixteen patients were randomized into each once-daily dosing group (10 mg, 25 mg, 50 mg, or placebo) evaluating oral SCH 417690 monotherapy for 14 days (four patients in each dosing group received placebo). All patients were either naïve to antiretroviral therapy or had been off treatment for at least eight weeks.
The drug was well tolerated and no adverse events were documented. After 14 days, HIV-RNA levels were 1.08 log10 copies/mL below baseline in the 10 mg group, 1.56 log10 copies/mL below baseline in the 25 mg group, and 1.62 log10 copies/mL below baseline in the 50 mg group. Approximately 45% of patients in both the 25 mg and 50 mg groups saw their viral loads drop by at least 1.5 log10 copies/mL.
ACTG 5211, a phase II study of vicriviroc with ritonavir and optimized background antiretrovirals in treatment-experienced patients, is actively enrolling patients.
GW 873140
Yet another CCR5 receptor antagonist in development is GlaxoSmithKline’s GW 873140. The drug has a long half-life and 50% occupancy of the CCR5 receptor persists five days after the last dose—with concurrent undetectable plasma drug levels—indicating that the drug likely has a lingering (“post-antiviral”) effect after it is stopped (Sparks, 2005). And like the other CCR5 antagonists, GW 873140 is a CYP 3A4 substrate. Coadministration with lopinavir/r results in a sixfold to eightfold increase in GW 873140 levels and a 30% increase in ritonavir levels (Adkison, 2005).
The results of a ten-day GW 874140 monotherapy study have been reported (Lalezari, 2004). The study enrolled 40 treatment-naïve and treatment-experienced HIV-infected subjects with R5-tropic virus, a CD4+ count above 200 cells/mm3, a viral load above 5,000 copies/mL, and had been off antiretroviral therapy for at least 12 weeks. The patients were randomized to one of four cohorts: 200 mg QD, 400 mg QD, 200 mg BID, or 600 mg BID. Each cohort had 10 subjects, with eight receiving active drug and two receiving matching placebo.
Twenty-one of the 40 patients were treatment-experienced and eight were coinfected with hepatitis C virus (HCV). Median baseline HIV-RNA levels ranged from 4.24 to 4.66 log10 copies/mL. Mean changes in HIV-RNA, after ten days of treatment, were -0.12 log10 in the placebo groups, -0.46 log10 among patients receiving 200 mg QD GW 874140, -1.23 log10 among patients receiving 200 mg BID, -1.03 log10 among patients receiving 400 mg QD, and -1.66 log10 among patients receiving 600 mg BID. The percentage of patients with a 1 log10 copies/mL drop or greater in viral load was 0% in the placebo group, 16.7% in the 200 mg QD group, 75% in the 200 mg BID group, 63% in the 400 mg QD group, and 100% in the 600 mg BID group. “Virologic suppression is similar to what we’re seeing with the other CCR5 antagonists,” Dr. Gulick said. “I think there’s a lot to look forward to with these three agents.”
The Concern of Coreceptor Switching
The three CCR5 antagonists are only expected to be active in people with R5-tropic virus. Studies have documented that, as HIV disease progresses, an increased percentage of patients have evidence of X4-tropic virus: HIV that is tropic for the CXCR4 receptor. Given that these agents do not work against X4-tropic virus, there’s some concern that R5 antagonists may result in the selection and proliferation of X4-tropic virus.
“What is the danger of this?” asked Dr. Gulick. “We really don’t now, but in people who have X4-tropic virus, there seems to be faster HIV disease progression. We’ve known this for more than ten years and there’s lingering concern about coreceptor switching in individuals receiving these drugs. In each of the CCR5 antagonist studies reported here, there has been evidence of coreceptor switching in one or two patients. Most, however, have switched back to R5-tropic virus when the compounds were withdrawn. Suffice it to say, the clinical implications aren’t yet clear.
AMD3100 and AMD070
AMD3100 and AMD070 are both CXCR4 antagonists from AnorMED Inc.. Because of toxicity concerns and limited efficacy, the development of AMD3100 was halted in May 2001. The company is now focused on AMD070, a compound that strongly inhibits viral infection by CXCR4-using virus—including virus using CXCR4 alone and/or virus using both CXCR4 and CCR5—in vitro. AMD070 is orally bioavailable in animals, shows additive or synergistic effects in vitro with other antiretroviral agents, and has yielded safe results in a recent Phase Ia dose-ranging study involving HIV-negative volunteers (Stone, 2004).
A Phase IIa study is being conducted by the Adult AIDS Clinical Trials Group (AACTG 5210).
| V. Integrase Inhibitors | Top of page |
The HIV integrase gene is essential for HIV replication and facilitates the integration of proviral HIV-DNA into the host cell genome. Unfortunately, it has not been easy to develop integrase inhibitors, despite the intense efforts of many investigators and many pharmaceutical companies. Challenges to development have included the lack of correlation of some integration inhibition assays with inhibition of whole virus replication, nonselectivity, adverse pharmacokinetic properties, and toxicity of many of the candidate compounds described to date.
The initial candidate integrase inhibitors were the diketobutanoic (“diketo”) acids, which work by sequestering the active divalent cation (Mg++) that is bound in the active site of the integrase gene by three acidic residues of the protein chain. Once the gene has been inhibited, the HIV-DNA forms inactive, unstable circular structures, and the virus is unable to replicate. Two earlier enzymatic functions of the integrase gene—assembly of pre-integration complexes and 3’ processing of the viral DNA ends—are not inhibited by diketo acids. They specifically inhibit the third step: strand transfer of viral DNA to cellular DNA. However, their pharmacokinetic properties did not lend themselves to drug development.
L-870,810
There are also the napthyridine carboxamides, a class with more favorable pharmacokinetic properties, which include Merck’s L-870,810. The results of a ten-day monotherapy study were reported at the 12th CROI (Little, 2005). A total of 30 HIV-infected patients, who were either antiretroviral-naïve or had been off of antiretroviral therapy for at least three months, were randomized to receive L-870,810 (200 mg or 400 mg BID) or placebo twice daily.
Seven patients received the 200 mg BID dose and 17 patients received the 400 mg BID dose. On day 10, at 200 mg BID, the mean decrease in HIV-RNA from baseline was 1.73 log10 copies/mL. At 400 mg BID, the mean decrease in HIV-RNA was 1.77 log10 copies/mL. L-870,810 at both doses was generally well tolerated with no discontinuation due to drug-related adverse events and no serious adverse events reported.
Unfortunately, the clinical development of this compound has been put on hold due to toxicity observed in dogs during long-term dosing. “Merck does have a backup compound that is going into clinical trials,” Dr. Gulick said. “We will definitely be hearing more about the integrase inhibitors in the future.”
| VI. Maturation Inhibitors | Top of page |
HIV assembly, maturation, and budding have long been focuses of drug development efforts. Maryland-based Panacos Pharmaceuticals has been developing PA-457, a “maturation inhibitor” that hinders a late step in gag processing involving conversion of the capsid precursor (p25) to mature capsid protein (p24) (Li, 2003). As a result, newly released viral particles are defective and non-infectious.
“PA-457 is a betulinic acid derivative,” Dr. Gulick commented. “It’s found in the bark of the London plane, or sycamore, tree. This should be an inexpensive compound to make, one would guess. But I don’t advise chopping down these trees just yet.”
Data from a phase I single-dose clinical trial involving HIV-infected patients were presented at the 12th CROI (Martin, 2005). Three doses were selected for evaluation: 75 mg, 100 mg, and 250 mg. At each dose level, six subjects received PA- 457; six additional subjects received placebo. The drug was well tolerated at all doses, with good oral bioavailability and favorable pharmacokinetics.
All three dose groups exhibited reductions in viral load compared with placebo that was sustained for more than 10 days in the higher dose groups. The median maximum viral load changes in the four groups were: –0.17 log10 copies/mL in the placebo group;–0.27 log10 copies/mL in the 75 mg PA-457 group; –0.45 log10 copies/mL in the 150 mg group, and –0.51 log10 copies/mL in the 250 mg group.
Additional studies of this compound are planned.
| References | Top of page |