
There is still considerable controversy over appropriate clinical management strategies for patients diagnosed with HIV during acute or early stage infection. However, it is widely accepted that enrolling longitudinal cohorts to study such patients is vital. Long-term outcome studies of persons enrolled early in the course of infection continue to yield important data regarding transmission dynamics, disease progression, efficacy of treatment, and incidence and consequence of superinfection. Not only are these studies helping to determine how best to clinically manage patients diagnosed early in the course of HIV disease, they are contributing significantly to the research exploring virologic, immunologic, and treatment questions in patients with chronic infection.
The Options Project, developed and conducted by the University of California, San Francisco (UCSF) AIDS Program at San Francisco General Hospital, is dedicated to the study of acute and early HIV infection. Dr. Frederick Hecht, a co-director of the program, and his colleagues continue to make headway in exploring various pathogenesis- and treatment-related issues affecting newly infected patients. In October, Dr. Hecht returned to PRN to provide members with an update on key data generated by the project, along with the recent findings from Acute Infection and Early Disease Research Program (AIEDRP), a national, multisite network consisting of research centers actively recruiting patients in the early stages of HIV infection. Here we present the highlights of his comprehensive talk.
| I. Treatment and Treatment Interruptions in Early HIV Infection | Top of page |
The interest in studies of structured treatment interruptions (STIs)-as a component of antiretroviral therapy started during acute or early HIV infection-dates back to a preliminary evaluation conducted by Dr. Eric Rosenberg and his colleagues at Massachusetts General Hospital (Rosenberg, 2000). This small study followed eight patients who began therapy during acute HIV infection and agreed to initiate STI after successful viral suppression, with the plan to restart therapy if viral load exceeded 5,000 copies/mL for three consecutive weeks or exceeded 50,000 copies/mL at any given time. Despite rebound in viremia, all eight patients were able to achieve at least a transient steady-state off therapy with viral loads below 5,000 copies/mL. At the time the report was prepared for publication in Nature, five out of eight subjects remained off therapy with viral loads less than 500 copies/mL after a median of six-and-a-half months. Dr. Rosenberg's group observed increased virus-specific CTLs and maintained HIV-specific CD4+ cells in all eight patients.
More recently, the Massachusetts General group reported extended follow-up data regarding six of the original subjects, along with eight additional patients who underwent a similar STI process (Kaufman, 2004). All 14 patients were HIV-antibody negative or in early seroconversion and had high HIV-RNA levels prior to beginning treatment (all patients received either a nelfinavir [Viracept]- or indinavir [Crixivan]-based regimen). Prior to initiating the first of up to four STIs, patients maintained HIV-RNA levels below 50 copies/mL for at least eight months while on treatment. And much like the eight-patient pilot study, patients were required to restart treatment if their viral load exceeded 5,000 copies/mL for three consecutive weeks or if they viral load exceeded 50,000 copies/mL at any one time.
The 14 patients in this STI study were followed for a median of 5.3 years. Eleven of the 14 (79%) patients were able to achieve viral loads of less than 5,000 copies/mL for at least 90 days following one, two, or three interruptions of treatment. However, a gradual increase in viremia and decline in CD4+ cell counts was observed in most individuals. By an intent-to-treat analysis, eight (57%), six (43%), and three (21%) of the 14 patients achieved a maximal period of control of 180, 360, and 720 days respectively, despite augmentation of HIV-specific CD4+ and CD8+ cell responses (see Figure 1). The magnitude of HIV-specific cellular immune responses before treatment interruption did not predict the duration of virologic control. The study authors also commented that the small sample size, as well as the lack of concurrent untreated controls, does not allow for the assessment of possible clinical benefit using this approach.
Figure 1. Mass General Cohort: Follow-Up of 14 Patients
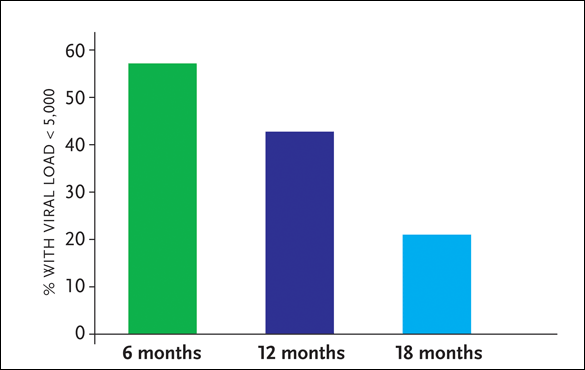
Source:Kaufman, 2004
“While these data suggest that STIs after treatment of primary HIV infection do not yield the level of dramatic control of viral replication suggested by the initial report, they raise some important questions,” Dr. Hecht said. “First, why isn’t viral control maintained? Second, is there a lower, but less dramatic difference in viral load set point? Figuring out what’s going on, even on a cellular level, could be valuable.”
| STI Data from the Options Project | Top of page |
To explore why viral suppression is lost, Dr. Hecht’s group turned to its own STI study conducted as a component of the Options Project. In this study, patients must have started antiretroviral therapy within six months of seroconversion, have remained on therapy for at least 24 weeks, and have achieved viral suppression (less than 50 copies/mL) for at least 16 weeks. If they meet these criteria, they are randomized to take one of two STI paths. The first path involves a main STI sequence in which treatment is resumed if the patient’s viral load is above 50,000 copies/mL at week 4, above 20,000 copies/mL at week 8, above 10,000 between weeks 9 and 24, above 200,000 copies/mL at any time point, or the CD4+ falls below 350 cells/mm3 at any time point. The second STI path involves a brief STI in which treatment is restarted if viral load is above 1,000 copies/mL for two consecutive weeks. Viral load must then remain below 50 copies/mL for eight weeks, followed by a main STI sequence.
New data were presented at the 2005 Keystone Symposium on HIV Pathogenesis in Banff, Canada (Schweighardt, 2005). At that time, Dr. Hecht’s group had evaluated six subjects who had completed the above protocol. Working closely with Dr. Douglas Nixon’s laboratory at the Gladstone Institute of Virology and Immunology, Dr. Hecht’s group attempted to characterize T-cell adaptive responses to HIV during treatment interruption in these six patients. The viral load of five of these patients peaked within two to four weeks off treatment, but then quickly declined, in some cases to less than 50 copies/mL.
The initial decline in viremia coincided with the development of high levels of gag-specific CD8+ cells of an effector phenotype. In all five subjects achieving low levels of viremia during early treatment interruption, there was viral rebound within 24 weeks. Viral mutations within the targeted immunodominant gag epitopes were detected at the time of viral rebound.
One STI patient that was unable to control viremia during STI had a very narrowly-directed immune response against two gag epitopes, elicited by CD8+ cells of a central memory phenotype. While Dr. Hecht’s group did not detect mutations within these gag epitopes during STI, it did detect multiple mutations in other regions of gag typically targeted by individuals with the same haplotypes. It was suspected that this patient was unable to control viremia due to the large number of immune escape mutations acquired before STI therapy.
These data indicate that STI can augment effective HIV-specific immune responses in infected individuals who initiate antiretroviral therapy during primary infection and that evasion of these immune responses by CTL escape may account for the inability to permanently suppress viral replication. “Putting all of this together,” Dr. Hecht added, “these data fit well with what we know about HIV biology outside of this kind of treatment setting. It looks as if we may be enhancing immune response to the virus. Using this approach, we may be maximizing the amount of [immune] pressure that the person’s immune system can put on the virus. How effective it is in controlling the virus depends on which epitopes are targeted. In some people, it may have some modest effect on the virus by forcing it to mutate more than it might otherwise, ultimately affecting its replication capacity. But a central question remains: are we actually achieving better results?”
| AIEDRP Observational Treatment Analysis | Top of page |
The Acute Infection and Early Disease Research Program (AIEDRP), a national, multisite network consisting of research centers actively recruiting patients in the early stages of HIV infection, is another source of important data regarding the use of antiretroviral therapy in acute/early disease.
In one study, reported earlier this year at the 12th Conference on Retroviruses and Opportunistic Infections (CROI) in Boston, Dr. Hecht and his AIEDRP colleagues reported on the outcomes of antiretroviral therapy for acute/early HIV infection after treatment discontinuation (Hecht, 2005). Subjects had to be enrolled in the AIEDRP cohort within six months of HIV seroconversion. They were offered combination antiretroviral therapy immediately upon entry. However, the decision to initiate therapy was voluntary and those who chose not to start therapy were followed in a manner identical to those who chose to start treatment.
A total of 58 patients were in the treatment group. They were required to have remained on therapy for at least 12 weeks and were then asked to stop treatment for at least four weeks. The data also included 337 patients enrolled in the cohort not receiving treatment. Primary outcomes were HIV-RNA levels and CD4+ cell counts at 24, 48, and 72 weeks, measured from the time of enrollment in the untreated group and after stopping antiretroviral therapy in the treatment group. The treatment group was divided into two subgroups based on when antiretroviral therapy was started: two or fewer weeks from seroconversion (acute infection) or greater than two weeks (early infection). Thirteen patients were in the treated acute infection group; 45 patients were in the treated early infection group.
The baseline characteristics were similar among the groups, although the patients with acute infection undergoing treatment had higher baseline viral loads (acute median log viral load = 5.8 log10 copies/mL in the acute HIV treatment group, 5.1 log10 copies/mL in the early HIV treatment group, and 4.9 in the untreated HIV group) and tended to have lower baseline CD4+ counts (488 cells/mm3, 527 cells/mm3, and 508 cells/mm3 respectively). Patients in the acute infection treatment group stopped their antiretrovirals after an average of 83 weeks of treatment. Patients in the early infection treatment group stopped their antiretrovirals after an average of 88 weeks of treatment.
According to the data presented, patients who started antiretroviral therapy within two weeks of seroconversion had viral load and CD4+ cell count benefits that were statistically significant for at least 18 months after stopping treatment. In contrast, persons who started antiretroviral therapy between two and 26 weeks after seroconversion had a viral load benefit at six months that was not sustained at later time points, and a CD4+ cell count benefit that appeared to decline after 12.
These data, Dr. Hecht indicated, suggest a prolonged benefit of antiretroviral therapy when initiated in acute HIV infection, and a more transitory benefit when treatment is initiated later in early infection. “Patients interested in early treatment should be informed of the risks, benefits, and uncertainties,” he said. “Enrolling patients into studies is certainly a worthwhile option to consider.”
| II. The Role of T-Cell Activation in Primary HIV Infection | Top of page |
The role of immune activation in HIV disease has been extensively evaluated in chronically infected patients. Study data support the hypothesis that HIV causes disease progression as a consequence of generalized T-cell activation and that the continuous high turnover of T-cells, along with the fact that the suppressed immune system is unable to regenerate new progenitor T-cells, eventually results in gradual loss of peripheral CD4+ T-cells. What has been missing, however, are data involving the role of immune activation in early HIV disease. This is an important piece of the puzzle, given that the immunologic and virologic events that occur during the earliest stages of infection can have a significant impact on subsequent disease progression.
To investigate the role of immune activation in early HIV disease, investigators associated with the Options Project—this time under the direction of Steven Deeks, MD—used the density of CD38 expression on CD4+ cells and CD8+ as a measure of T-cell activation in 68 antiretroviral-untreated and 83 antiretroviral-treated acutely and recently infected adults (Deeks, 2004).
Results of the study indicate that, at study entry, T-cell activation was positively associated with viremia, with CD8+ cell activation levels increasing exponentially at HIV-RNA levels greater than 10,000 copies/mL. Among untreated patients, the level of CD8+ cell activation varied widely between individuals but often remained stable within a given individual. CD8+ cell activation and plasma HIV RNA levels over time were independently associated with the rate of CD4+ cell loss in untreated individuals.
Based on these data, a number of observations were made. “First,” the authors write in the paper they published in Blood last year, “there is a strong and consistent relationship between the level of viremia and the level of both CD4+ and CD8+ cell activation during acute HIV infection. Second, most untreated patients reach a steady-state level of T-cell activation in early HIV infection. This immunologic activation “set-point” varies widely between individuals but is generally stable within a given individual. Third, the CD8+ cell activation “set-point” during untreated HIV infection is a strong independent predictor of the rate of CD4+ cell decline. Fourth, initiation of antiretroviral therapy during early HIV infection dramatically reduces the level of CD8+ cell activation and, to a lesser degree, CD4+ cell activation.”
Collectively, these data support the concept that the course of HIV disease is not only dependent on the actual level of viral replication—the viral load “steady state”—but also on the ability of the virus to cause sustained increases in CD8+ cell activation. Like many other relationships between HIV and the host, the association between the virus and the degree of immune activation is established early in the course of HIV infection.
Might it be useful to limit immune activation in acute/early HIV infection? This is an important question that is being explored in an AIEDRP study (AI 501) using cyclosporine A.
Cyclosporine A (CsA) is an immunosuppressant that modulates the activation of CD4+ cells. Specifically, CsA binds to cyclophylin (CpN) in the cytoplasm of cells, forming a complex between CsA and CpN. This CsA/CpN complex binds to and blocks the function of the enzyme calcineurin (CaN), which in turn blocks the transport of the nuclear factor of activated T-cells (NF-ATc) to the cell’s nucleus. If NF-ATc does not reach the nucleus, it cannot bind with the nuclear component of the nuclear factor of activated T cells (NF-ATn). It is this NF-ATc/NF-ATn complex that is necessary to initiate interleukin-2 (IL-2) production. Without sufficient IL-2 production, CD4+ cells will not be activated, potentially reducing immune activation and the proliferation of viral replication.
The hypothesis supporting AI 501 is that CsA, given with antiretroviral therapy during acute HIV infection, will reduce the size of the latent pool of HIV-infected cells and preserve both HIV-specific and non-HIV-specific immunity due to sparing of CD4+ cells by preventing activation and infection. Approximately 50 patients will be randomized in a 2:1 fashion. Patients must be EIA negative or present with three or fewer bands on a Western blot. Patients will also need to have HIV-RNA levels in excess of 500,000 copies/mL. All patients will receive lopinavir/ritonavir (Kaletra) plus abacavir/zidovudine/lamivudine (Trizivir). Two-thirds of the patients will also receive CsA, 0.3 mg/kg twice daily for four weeks. The primary endpoint is the level of proviral DNA at 48 weeks and the secondary endpoints include absolute CD4+ cell counts at various time points and measures of HIV-specific immunity.
There are ample data to back the rationale of AI 501. In one study, the virologic and immunologic effects of immunomodulation during primary simian immunodeficiency virus (SIV) infection were examined in monkeys (Martin, 1997). The animals were treated with either cyclosporine or placebo for 32 days, beginning five days before SIV inoculation. Duration of antigenemia decreased in 5/7 treated monkeys, with two monkeys experiencing delayed onset and lower peak antigenemia. Proviral DNA levels in blood and lymph nodes, along with infected cell numbers in lymph nodes, were also transiently decreased. What’s more, transient increases in CD4+ cell counts were seen in the cyclosporine-treated monkeys, compared to those in the control group.
Data are also available from a study involving humans with acute HIV infection (Rizzardi, 2002). In this study, a Swiss team evaluated the safety and the immune-modulating effects of combining CsA (0.3 to 0.6 mg/kg every 12 hours) with antiretroviral therapy in nine patients with primary HIV infection. After eight weeks of treatment, all patients discontinued cyclosporine but remained on antiretroviral therapy.
Viral replication was suppressed to a comparable extent in the CsA-treated patients and in 29 control patients whose primary infection was treated with antiretroviral therapy alone. No differences in cell-associated DNA or RNA were seen in the two groups. Patients in the CsA group experienced significantly higher CD4+ cell counts, both during the initial eight-week regimen and up to a year later. What’s more, patients in the CsA group had higher HIV-specific CD4+CCR7- cells—effector memory cells—than those treated with antiretroviral therapy alone.
| III. HIV Superinfection | Top of page |
HIV superinfection—sequential infection with variants of HIV—has long been a source of debate and concern among clinicians and public health officials. The topic is of particular interest to researchers involving in the study of primary HIV infection, given that the five case reports published to date have involving individuals who were recently infected or intermittently treated with antiretroviral therapy (Ramos, 2002; Jost, 2002; Altfeld, 2002; Koelsch, 2003; Smith 2004; Brenner, 2004). Studies of chronically infected patients have not shown evidence of superinfection (Gonzales, 2003; Grant, 2004; Tsui, 2004).
“The fact that we’ve only seen case reports involving newly infected patients suggests that superinfection can occur before development of protective immunity,” Dr. Hecht commented. “Understanding superinfection may be a key to understanding protective immunity and vaccine development. And it most definitely raises patient counseling issues.”
At the 12th CROI in Boston, Dr. Hecht’s group—which included Dr. Robert Grant and his colleagues with the UCSF Laboratory of Clinical Virology—presented data on the frequency of sequentially expressed dual HIV infections (SEDIs) in the Options Project Cohort (Grant, 2005).
Dr. Grant’s lab evaluated samples collected from all patients enrolled in the cohort who had been followed for at least 48 weeks and had remained off treatment. “We first looked at the pol gene—using the sequencing performed when looking for drug resistance—and compared their baseline specimen with their most recent available follow-up specimen,” Dr. Hecht explained. “We conducted phylogenetic analyses to see if the subsequent viruses matched up with the viruses they first had. When it looked like we had a possible superinfection case, we did a lot of retesting with specimens throughout the entire series of time points to see if we could support this finding and to look for exactly when superinfection occurred.” Finally, to evaluate population dynamics, Dr. Hecht’s group used heteroduplex assays to determine if the second virus was evident in the baseline specimens (this method has been validated at 1.5% to 3.0% sensitivity for minor variants). “We can get down to pretty low quantities using these methods to look for evidence of the second virus at early time points.”
A total of 104 recently infected individuals were analyzed at two or more time points, representing 195 person-years of observation. All viral sequences were subtype B. Highly divergent viral sequences appeared in a total of four cases over time, representing an incidence density of 2.1/100 person years. “This incidence is close to what you might expect in a reasonably high-risk cohort in San Francisco of HIV-negative individuals becoming infected with HIV,” Dr. Hecht said. The heteroduplex assays confirmed the lack of second virus at baseline, indicating that these were sequential infections.
One patient discussed by Dr. Hecht—provided with the pseudonym “Hector”—saw a highly divergent virus appear 16 to 44 weeks after infection (see Figure 2). “Looking at his HIV-RNA levels, he appeared to be doing very well,” Dr. Hecht pointed out. “Around week 24, his viral load leveled off at approximately 100 copies/mL. Then, around week 40, there was a sharp increase in his viral load, spiking at around 10,000 copies/mL. It was here that we noted that he had a different virus than the one he started off with. His CD4+ cell count, which was initially doing well, started to decline rapidly. In fact, he had to go on treatment within another few months of follow-up.”
Figure 2. "Hector"
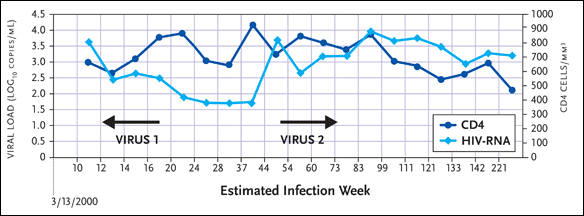
Source:Schweighardt, 2005
Another interesting case is “Cody” (see Figure 3). A sharp increase in his viral load occurred between weeks 43 and 53, before his second virus was detected. In this patient, multiple-drug-resistant virus was replaced by wild-type virus, which was associated with more rapid disease progression. His CD4+ count, reaching a high of approximately 900 cells/mm3 at week 35, fell to a low of approximately 300 cells/mm3 at week 79.
Figure 3. "Cody": Replacement of MDR HIV with Wild-Type HIV
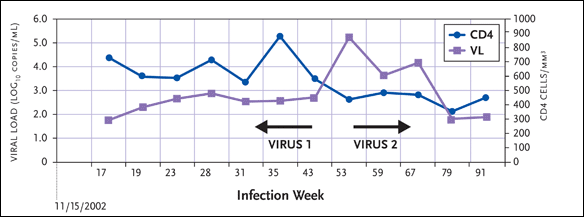
Source:Schweighardt, 2005
With “Jonah,” the second virus appeared concomitantly with a diagnosis of gonorrhea, at week 168 (see Figure 4). “His viral load spiked at 10,000 copies/mL at the time of the gonorrhea diagnosis and his CD4+ cell count began a rapid decline after this time point.”
Figure 4. "Jonah": Virus 2 Appears with Diagnosis of Gonorrhea
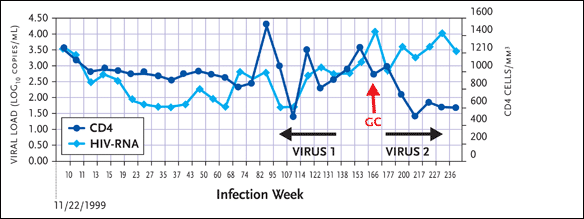
Source:Schweighardt, 2005
“Leon’s” first virus was detected through to week 29. His second virus was first detected at week 39. Around the time his second virus was detected, his CD4+ count was approximately 700 cells/mm3. Around week 48, his CD4+ count plummeted to 350 cells/mm3. After a transient increase, his CD4+ count fell below 100 cells/mm3 around week 100. “With this patient, the second virus appeared before rapid progression to AIDS.”
“All four patients saw increases in their viral loads with their second viruses,” Dr. Hecht said. “The average increase was 1.4 log10 copies/mL, compared to an average decrease of 0.24 log10 copies/mL in our untreated cohort participants without superinfection.”
In summarizing these data, Dr. Hecht remarked that superinfection appears to be relatively common in early HIV and may be associated with increased viral load and more rapid disease progression. “What we’re interested in finding out about now is the closing of the superinfection window. Why do people stop becoming superinfected, at least from what we think we’re seeing, after the first few years of infection? This question remains unanswered.”
| References | Top of page |